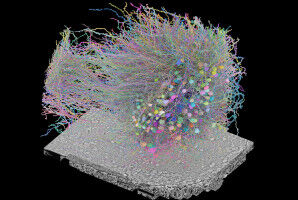
Hydration restores the airway surface seal. In turquoise, the junctions between the epithelial cells (blue).
Hydration restores the airway surface seal. In turquoise, the junctions between the epithelial cells (blue).
Hydration restores the airway surface seal. In turquoise, the junctions between the epithelial cells ( blue ). UNIGE - Laboratory of Prof. Marc Chanson - A team from the University of Geneva reveals that hydrating the surface of the airways of people with cystic fibrosis restores their protective barrier against unwanted bacteria. Cystic fibrosis is a rare genetic disease which can cause very serious symptoms. In particular, patients suffer from chronic bacterial infections that can lead to respiratory failure. It is caused by mutations in the CFTR gene, which regulates water movement across the cell membrane. Consequently, mucus quality is altered, it is no longer capable of capturing undesirable bacteria and expelling them.
UM DIESEN ARTIKEL ZU LESEN, ERSTELLEN SIE IHR KONTO
Und verlängern Sie Ihre Lektüre, kostenlos und unverbindlich.
Ihre Vorteile
- Zugang zu allen Inhalten
- Erhalten Sie Newsmails für Neuigkeiten und Jobs
- Anzeigen veröffentlichen